Treephyler: fast taxonomic profiling of metagenomes.
Assessment of phylogenetic diversity is a key element to the analysis of microbial communities. Tools are needed to handle next-generation sequencing data and to cope with the computational complexity of large-scale studies. Here, we present Treephyler, a tool for fast taxonomic profiling of metagenomes. It combines the predictive power of tree-based and speed of signature-based approaches. Treephyler was evaluated on a real metagenome to assess its performance in comparison to previous approaches for taxonomic profiling.
How to cite
F. Schreiber, P. Gumrich , R. Daniel and P. Meinicke (2010)
Treephyler: fast taxonomic profiling of metagenomes
Bioinformatics 26, 960-961
News
- March 2011: Treephyler now supports Hmmer version 3.0 and the new Comet format
Download
- Treephyler source code: download (.tar, 120 Kb)
- Readme: download (.txt, 42 Kb)
- Glacier dataset: website (@NCBI, 277 Mb)
- Carma predictions : download (.tar, 12 Mb)
- treephyler predictions: download (.tar, 12Mb)
- phymm predictions: download (.tar, 138 Mb)
- Glacier dataset (6-frame translated): download (.tar, 468 Mb) with UFO predictions: download (.tar, 70 Mb)
The tree parsing method
We use the method of Nguyen et al [1] to parse classify the query sequences in a phylogenetic tree.
The algorithm traverses a tree and assigns query sequences to a taxonomic rank of at least three reference sequences in the same subtree belong to the same
taxonomic rank. The query sequence will then be assigned to the lowest taxonomic rank all reference sequences have in common.
In case there is no such overlap, the query sequences will not be assigned to a taxonomic rank.
Evaluation
Dataset
The Glacier Ice Metagenome Of The Northern Schneeferner (Info)
Glacier ice samples were collected in June 2005 at the Northern Schneeferner (47° 25′ N, 10° 59′ E), which is located on the Zugspitzplatt (Germany).
In order to avoid contamination by surface melt water, the first 30 cm of glacier ice were removed and discarded. Ice up to a depth of approximately 0.5 m was collected and transported frozen to the laboratory.
To isolate DNA the ice was melted at 4°C, and portions of 1 to 2 litres were filtered using a sterile filter unit with a cellulose acetate membrane (pore size 0.2 µm; Whatman, Dassel, Germany).
The cell-containing membrane filters were used as starting material for DNA isolation (for details see [2]).
The glacial DNA was sequenced by conducting two full runs (70x75 picotitre plates) on a Roche GS-FLX pyrosequencer (Roche, Mannheim, Germany).
The preparation of DNA for pyrosequencing and pyrosequencing were done according to the manufacturer's protocols (Roche).
Pyrosequencing of this DNA yielded 1,076,539 reads with an average read length of 223 bp.
Methods
- Treephyler
- Carma [3]
- Phymm [4]
- 16S RNA analysis
Runtime comparison - treephyler vs. other tools
We randomly chose 1% = 10,765 sequences of the glacier dataset to get results for all tools in a reasonable timeframe.
All analyses were carried out on one core of a dual-core AMD opteron 2216 2,4 GHz and 16 Gb RAM. "Execution time 100%" means that we conducted the analysis using the complete
glacier ice dataset. Due to the computational complexitiy of Carma, the runtime of Carma was interpolated.
| Method | No. of cores | Execution time 1% | Execution time 100% |
|---|
| Treephyler |
1 |
0,3 h |
12 h |
| Phymm |
1 |
0,3 h |
30 h |
| Carma |
1 |
168,1 h |
696 h |
Runtime comparison - treephyler
We used the full glacier ice dataset to conduct runtime analyses on varying number of computers.
The first analysis were done on our computer cluster using 50 processor cores (AMD opteron 2216 2,4 GHz, 8 Gb RAM), the second analysis was done
on a single computer with 8-cores.
| Method | No. of cores | Execution time / core | Percentage of assigned reads |
|---|
| Treephyler | 50 | 13 mins | 15% |
| Treephyler | 8 | 139 mins | 15% |
Prediction comparison
The assessment of accuracy of the different methods is based on the full glacier ice dataset (1,076,539 sequences).
Treephyler and Phymm were run on own computers, whereas the results of Carma were taken from 3.
| Method | Percentage of assigned reads |
|---|
| Treephyler | 15% |
| Phymm | ~99 % |
| Carma | 15% |
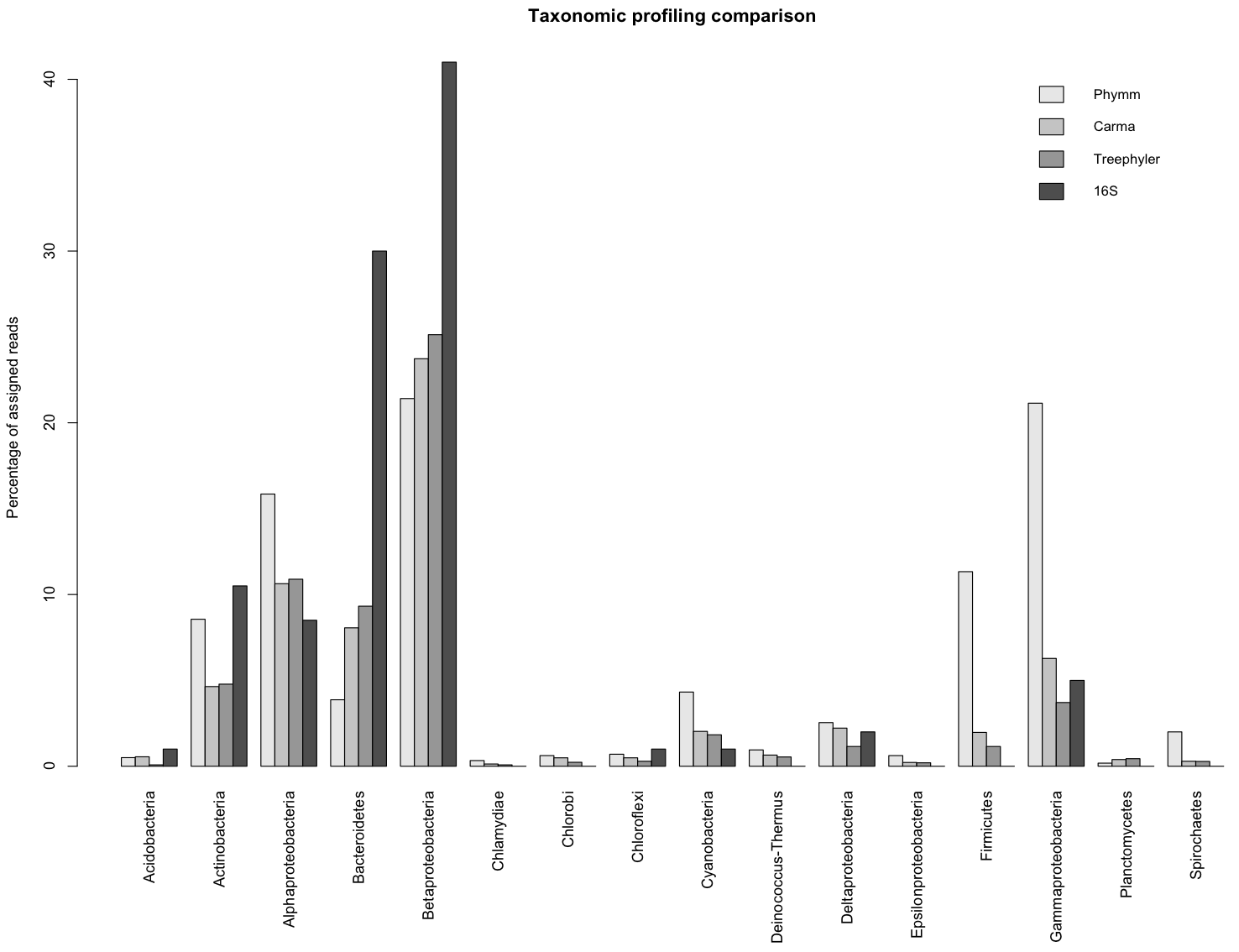
Phylum level
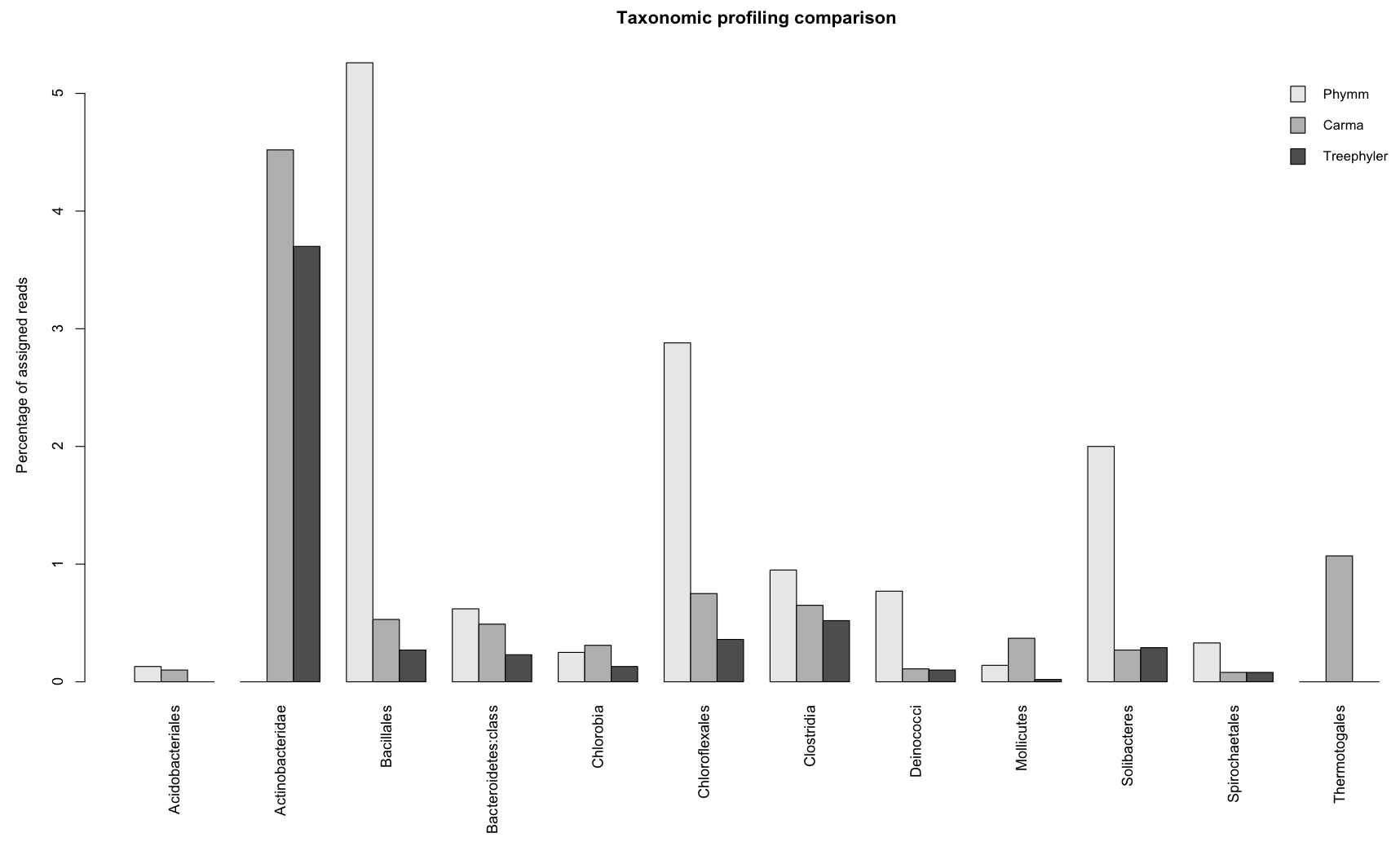
Class level
Additional comparison were performed on the class level for bacteria. Since there were no predictions available for the 16S analysis, only
the three methods Treephyler, Carma, and Phymm could be compared.
Please direct your questions and comments to fabian@gobics.de.
References:
[1] Nguyen, T.X. et al. (2006) Phylogenetic analysis of general bacterial porins: a phylogenomic case study, J Mol Microbiol Biotechnol, 11, 291-301.
[2] Simon C, Herath J, Rockstroh S, Daniel R (2009) Rapid identification of genes encoding DNA polymerases by function-based screening of metagenomic libraries derived from glacial ice. Appl. Environ. Microbiol. 75: 2964-2968.
[3] Krause, L., Diaz, N.N., Goesmann, A., Kelley, S., Nattkemper, T.W., Rohwer, F., Edwards, R.A. and Stoye, J. (2008) Phylogenetic classification of short environmental DNA fragments, Nucleic Acids Res, 36, 2230-2239.
[4] Brady, A. and Salzberg, S.L. (2009) Phymm and PhymmBL: metagenomic phylogenetic classification with interpolated Markov models, Nat Methods, 6, 673-676.