Phylogenetic Sequence Analysis
Fabian Schreiber
Exercises
A common task nowadays is to classify a newly discovered protein. To classify a new protein we have to relate it to other (already known) proteins. After the collection of related (homologous) proteins (Homology Search), the sequences are related to each other (Alignment) and some criteria (e.g. Neighbor-Joining, Maximum Likelihood) is used to calculate the degree of relatedness between the proteins. The results will be printed as a phylogenetic tree.The focus of this exercise is to get familiar with programs to use for the different steps rather than to answer a specific biological question. Unless told otherwise, please leave all settings on their default values.
I. Homology Search
In this question, we will search for homologous sequences for a given nucleotid sequence.
|
1.
1.Open the browser and go to Genbank.
Get the complete coding sequence for the human phosphoprotein p53 gene. Choose Genbank (full) as a display mode.
The right sequence is 2944 base pairs long and has the accession number AH002918.1
Sequence: download
2. Go to BLAST. Since BLAST is a collection of different methods for searching for similar sequences, choose the right method (under Basic BLAST). Perform a BLAST search of this sequence against the full non-redundant nucleotide database, restricting your results to Mammalia (by entering Mammalia[organism]) in the Entrez Query field).
3.
Wait until the search has finished and take a close look on the result page as well as on the taxonomy page to get an overview.
BLAST Result: download
Taxonomy Report: download
4.
On the result page , you will see hits from many different species (the species are named next to each hit in the table of results). Choose 8 of the species which appear in the results (including human).
Hint: use the taxonomy page to find the best hit/species.
5. Open each of the 8 hits in a new tab/window. You will see the corresponding entry of the nucleotide database in genbank format with additional information (e.g. Organism, Reference, Length). Since most alignment and phylogeny programs cannot handle the genbank format, display the entry in fasta format.
6.
Open a Texteditor (e.g. Kate, emacs, vi/vim, etc) to store all 8 sequences consecutively.
Sequences: download
Sequences(altered fasta header): download
II. Multiples Sequence Alignment
To bring the sequences from task 1 in relation to each other you need to compute an alignment. 
|
1. Go the the ClustalW server at European Bioinformatics Institute.
2. Enter all your sequences together in the provided field and click Run.
3. Your results will appear within a few seconds within the web browser.
4. Now view your alignment by clicking alignment file.
5.
Store the alignment in a text editor.
Sequence Alignment: download
III. Building a phylogenetic tree
There are different methods to construct a phylogenetic tree from a multiple sequence alignment. In this question, we will create two phylogenetic trees using Neighbor-Joining and Maximum Likelihood methods. 
|
Neighbor-Joining:
1. Go to the online Phylip page.
2. Click dnadist under Programs for molecular sequence data.
3. Enter your email address and the results of the alignment in the correct fields.
4. Click Run dnadist.
5.
Once the web page appears you can view the constructed distance matrix in outfile.
Distance Matrix: download
6. To apply the neighbor joining select neighbor the menu on the web page and click Run the selected program on outfile.
7. Click Run neighbor in the next page and then wait for the results to appear.
8.
Click outtree on the results page to get a description of the phylogenetic tree. Save the tree in a text editor.
Tree: download
Maximum Likelihood:
9. Now go back to the online Phylip page and click fastDNAml.
10. Enter your email address and the alignment results again and click Run fastdnaml.
11.
Once the results page appears, click treefile to get a description of the phylogenetic tree and save it in your text editor. Make sure you keep track of which method created which tree.
If you're interested, the format for tree descriptions is described here.
Tree: download
IV. Viewing a Tree
In this question, we will take the trees you created and turn them into human-readable pictures, then analyze those pictures.1. Go to PHYFI, a viewer for phylogenetic trees, and copy one of the trees in the corresponding field.
2.
Click Draw to view the tree in a human-readable picture.
Distance-Tree: download
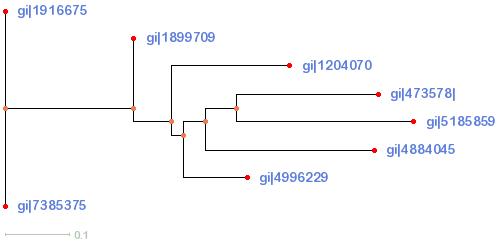{kind=link}
3.
Do the same for the other tree.
Likelihood-Tree: download
{kind=link}
4. On each tree, label each node with the name of its corresponding species (on a sheet of paper). You can find the species names that correspond to the accession numbers in the multiple sequence file.
5. Explain briefly two of the differences you can see between the phylogenies you obtained.
6.
On your taxonomy printout from question 1, draw lines on the left between the 8 species you marked to imitate a phylogenetic tree. Take careful note of the hierarchical layout of the report.
The easiest way to do this is to start short horizontal lines going left from each of the 8 species. At the earliest level where two or more species have a common classification, join their lines with a vertical branch and begin a new horizontal branch in the middle of that to continue extending left.
7.
Choose one of the phylogenetic trees that Phylip generated and explain briefly two of the differences between it and the quasi-phylogenetic tree you just drew.
If there are less than two differences, say so.
8.
Give a reason why the hierarchy in the taxonomy printout might not accurately represent evolutionary history.
Think about how the traditional taxonomical classification was made.