University of Göttingen - Faculty of Biology - Institute of Microbiology and Genetics - Department of Bioinformatics
Methods Course: Gene Prediction
Mario Stanke
Presentation: pdf
Exercises:
1) Download the human DNA sequence HSHAP to your computer and save it in a file called HSHAP.fa. The suffix '.fa' stands for the fasta format. Use the web interface of the program AUGUSTUS at augustus.gobics.de to predict the gene structure of this sequence. Use the standard settings of the options of the web interface. Compare the prediction with the true gene structure in text format or as a picture.
2) Predict the gene structure of the same DNA sequence using the program Genscan. Again, compare with the true gene structure. What is wrong?
3) Now try an extrinsic method. First make a blastx search of HSHAP
to the nr protein database by starting from the NCBI
BLAST page. From the resulting protein hits take the top ranking protein
hit and create a fasta file containing the amino acid sequence of this
protein. Only if BLASTX takes too long, take this prepared protein
sequence.
Then use Genomescan
for another prediction of the gene structure of that DNA sequence HSHAP.
Genomescan is like Genscan but uses in addition to the input DNA sequence
a protein sequence. This protein should be similar to one of the protein sequences of the genes encoded in the input DNA
sequence. Use the protein sequence you found in the BLASTX search. What
does Genomescan predict? How could the informant protein sequence could
have helped?
4) We have the expressed sequence tag (EST) BU168206 and want to find the gene
whose cDNA that EST is part of. The EST is only a part of the complete cDNA. Suppose we have already localized a human
genomic sequence that seems to contain that gene (e.g. using blastn).
We want to predict the genes in that genomic sequence using the information that the EST BU168206 can give us.
Make a spliced alignment between this EST and the genomic sequence using the program
sim4. What conclusions do you draw from the spliced alignment of sim4?
How can you pin down a part of the gene structure of the gene?
Use the expert options of AUGUSTUS and specify contstraints that force AUGUSTUS to predict a gene structure that
is compatible with the information you derived from the EST alignment.
Just in case: sim4 output (Don't cheat!)
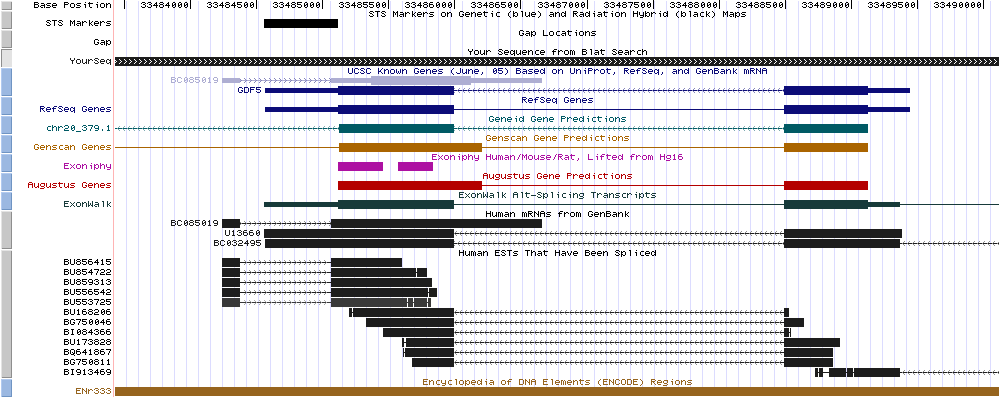
5) Use BLAT to find the region on the human genome from where I have taken the sequence ENr333.part.fa from the previous exercise. On the UCSC genome browser zoom into the region of ENr333.part.fa containing the annotated gene GDF5. Switch spliced ESTs to full display, swich the display of the AUGUSTUS genes on and include a couple of more gene predictions (e.g. Genscan, Geneid). Your picture should look like this or like that.
6) Try a comparative gene prediction approach to predict the genes in this syntenic human/mouse sequence pair. human: L15533.fa, mouse: D63360.fa. This is the sequence pair from the picture in the talk. Point your browser to the AGenDA web server and let it predict the gene structure of these two sequences. Make sure to select the correct groups of species at the top of the form. You can compare the predictions of AGenDA with the annotations in the genbank files L15533.gb D63360.gb.
7)
Download the executable program AUGUSTUS from the AUGUSTUS web page
and run it locally on the sequence ENr333.part.fa. Either follow my intructions
with the projector or try to do it on your own:
First save the file augustus.1.7.src.tar.gz in your home directory.
Then unpack it by typing
tar xzf augustus.1.7.src.tar.gz
on the command line of a terminal window. Then read the README file in the augustus
directory and follow the instructions. We can make the program output alternative gene structures. These alternatives
are less likely than the standard prediction according to the model but are still plausible and could be correct.
Do this by running AUGUSTUS on the command line like this
augustus --species=human ENr333.part.fa --alternatives=true
How many alternative transcripts does it predict for the gene containing the EST? And how do they differ from each other?
{kind=link}
{kind=link}